How to Use

Perception of taste is a complex sensation evolved in humans primarily to respond to naturally occurring food-derived chemicals. Among all the taste perceptions, the dichotomy of sweet and bitter tastes is of special importance to human gustatory mechanisms. The sweetness is innately attractive, whereas bitterness evokes an aversive response. A better understanding of molecular correlates that are responsible for a gradient of bitter-sweet taste is of key value towards the identification of natural as well as synthetic compounds of desirable taste on this axis. Towards this goal, an extensive compilation of bitter and sweet molecules is a necessary prerequisite to build suitable computational models for addressing this problem.
BitterSweet is an extensively curated database consisting of over 25,000 molecules obtained from ten different resources which are divided into three taste categories: Bitter, Sweet or Tasteless. Apart from the repository of bitter and sweet molecules, it also provides a tool for predicting the bitter and sweet probability of an arbitrary compound based on a state of the art machine learning model designed for classification of bitter and sweet molecules using open source chemical descriptors. The database is further enriched with predicted bitter-sweet probabilities for molecules from external datasets such as FlavorDB Foodb and DrugBank, in addition to the curated molecules.
BitterSweet database enables insights into the perception of bitter and sweet tastes as well as informed leads for engineering new molecules of desired taste gradient. Along with the power of computational strategies for spanning the chemical space of known and unknown realizable organic compounds, this resource presents with a potent tool for generating compounds of desirable bitter-sweet potency. In the presence of an epidemic of nutrition-related diseases such as obesity and diabetes, BitterSweet facilitates the search for molecules that can increase the appeal of food and beverages to address the problem of overnutrition.
BitterSweet Search
Search by Molecule ID
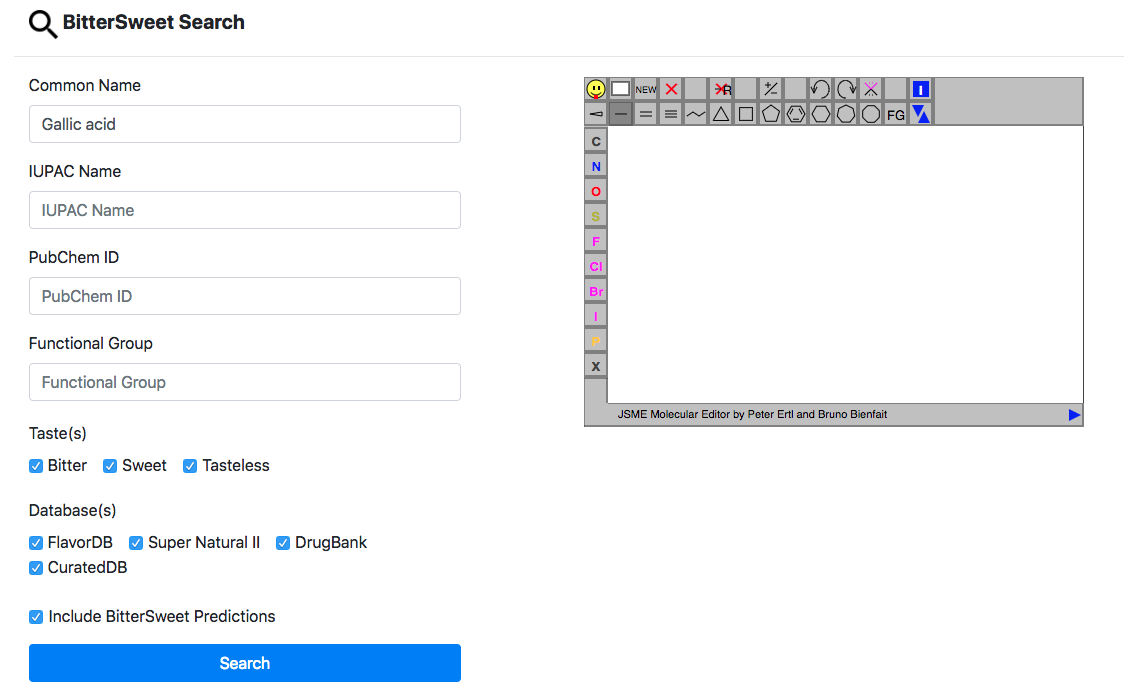
This the primary search tool for querying and browsing bitter, sweet and tasteless
molecules curated from a large number of sources. In addition, we provide search for
molecules from external databases for which probability of bitter and sweet taste has
been
predicted using a state of the art model. The search could be performed using a range
of
chemical descriptors namely Common Name, IUPAC Name, PubChem ID and functional group.
The
conjunction of query parameters allows a refined search using multiple parameters.
Searching without any query will yield all the molecules present in the selected
sources.
Note: An empty query will result in listing of all the molecules present in the
database.
If you wish to check out the list of all the molecules in BitterSweet, please Click Here. You can further query for molecules present in the individual databases/type by simply selecting them and leaving all other queries blank, for example -
- All the molecules from FlavorDB can be found here
- All the molecules from CuratedDB can be found here
- All bitter molecules can be found here
- All sweet molecules can be found here
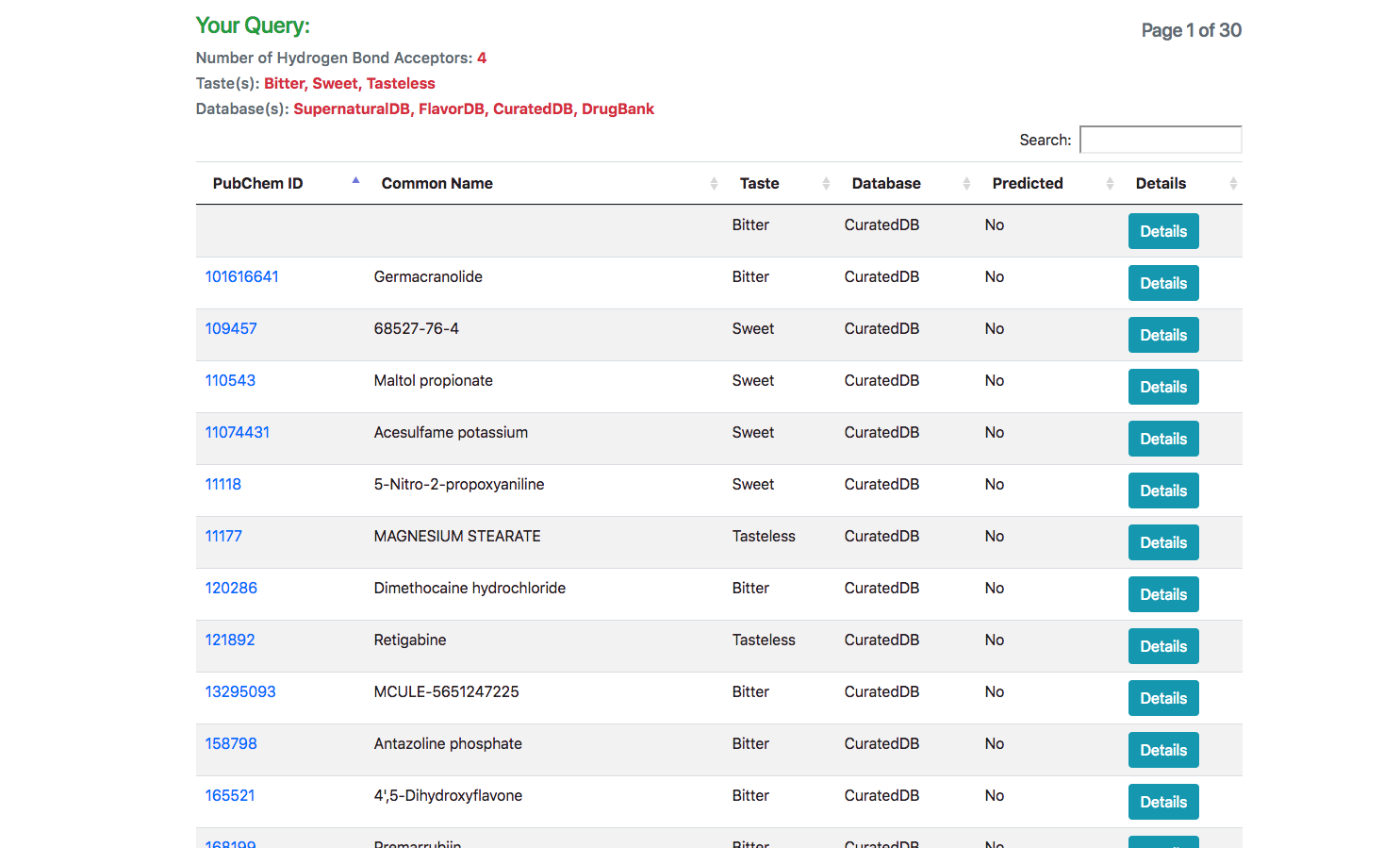
Search Results
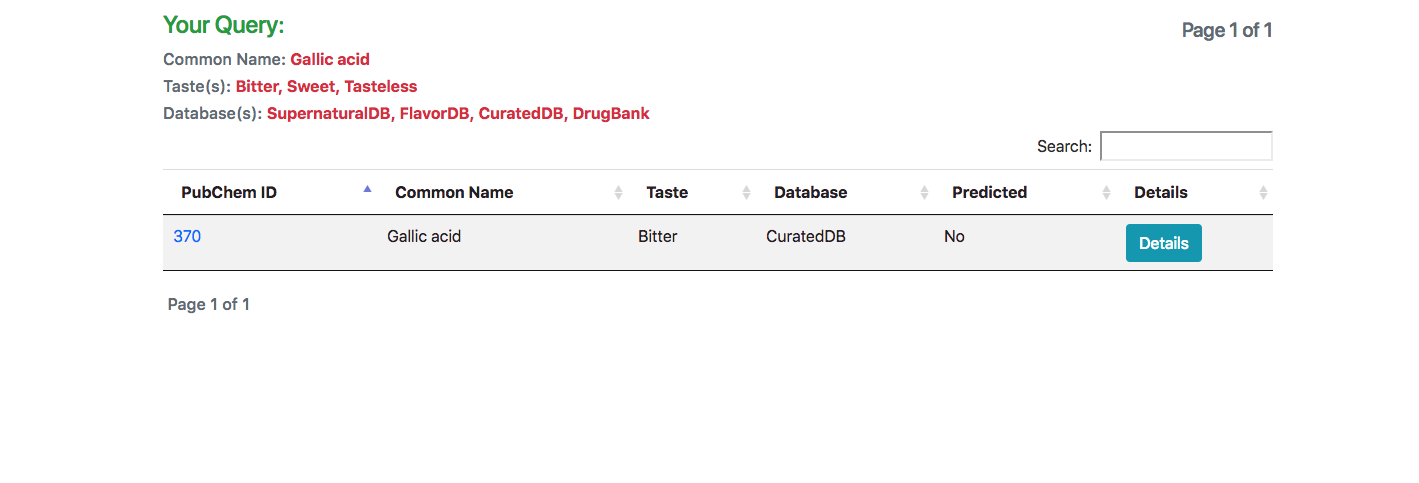
In response to the query, a list of all matching molecules is listed. In case of using the JSME tool for searching using a structure, an additional column indicating the similarity of the molecule is also displayed.
Information Panel
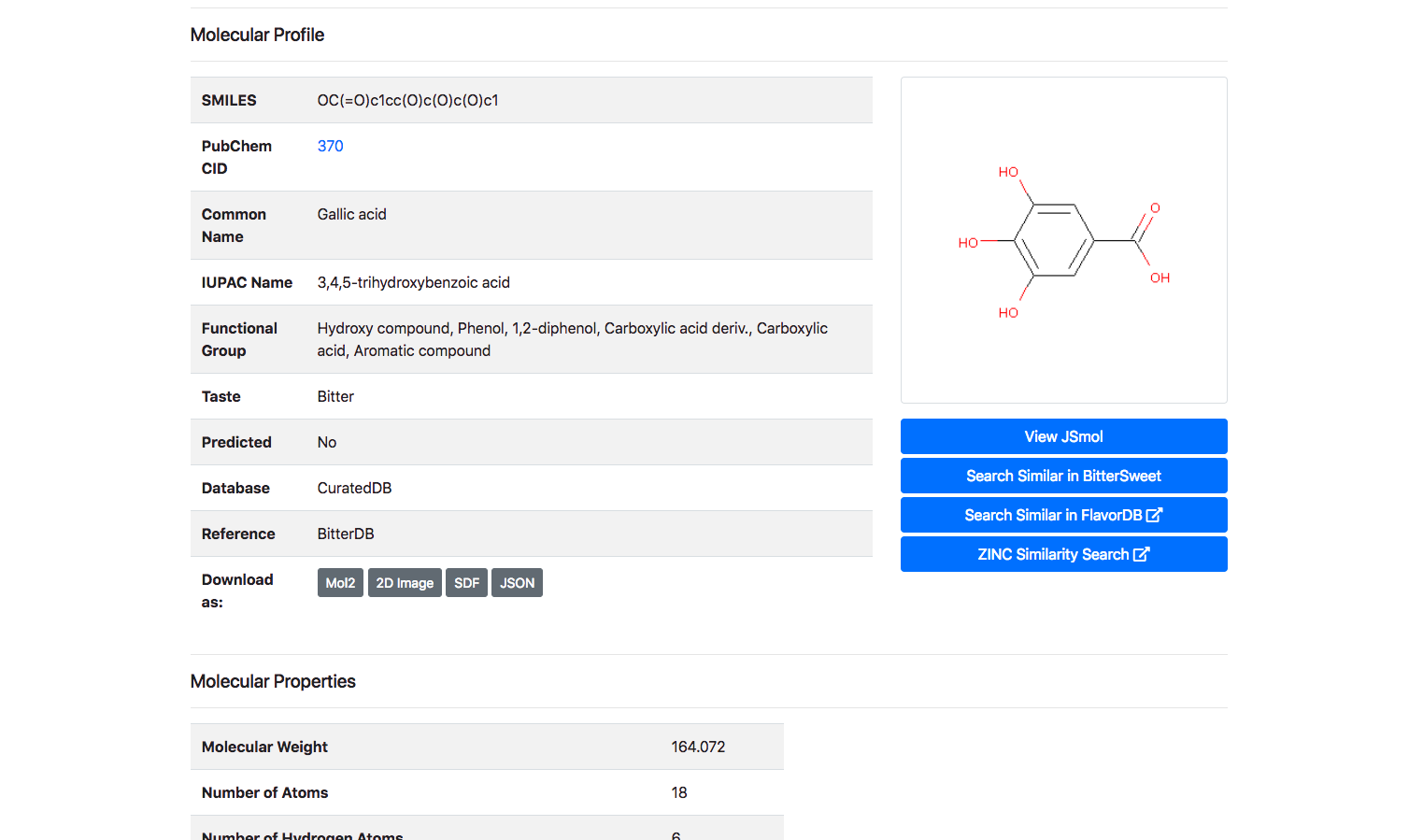
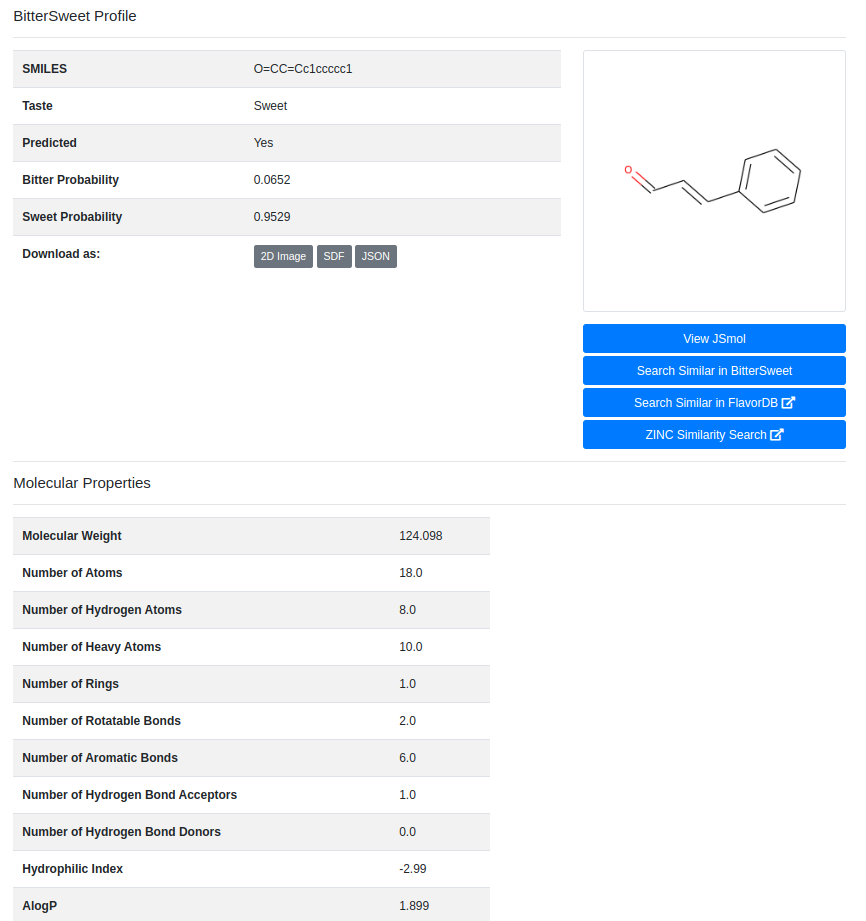
Upon clicking the “Detail” button against a molecule, the user is routed to a page with its complete entire. This includes details of chemical identifiers such as Chemical Structure, Common Name, PubChem ID, SMILE ID, IUPAC Name, Functional Group details, Taste, Source Database and Reference. In addition to this chemical characteristics such as Molecular Weight, Number of Atoms, Number of Hydrogen Atoms, Number of Aromatic Bonds etc are provided. The molecule could be downloaded as a Mol2, SDF, JSON or a 2D Image.
Advanced Search
Search by Chemical Properties
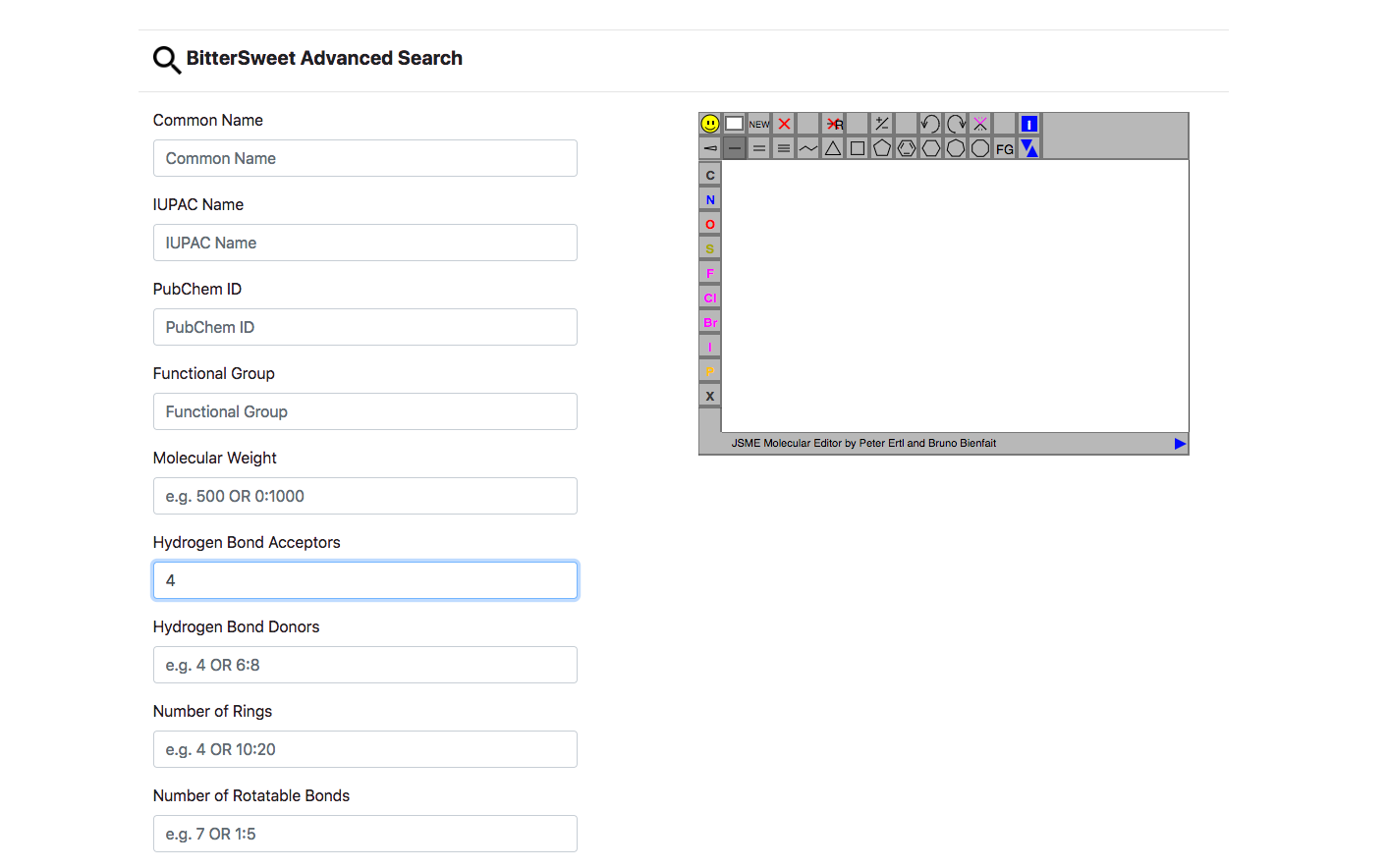
Beyond the basic search, Advanced Search enables queries via other descriptors such as Molecular Weight, ALogP, Hydrogen Bond Acceptors/Donors, Number of Rings, Number of Rotatable Bonds, Number of Aromatic Bonds, AloP etc. Similar to the basic search Advanced Search allows filtering by conjunction of parameters, including a graphic query via its chemical structure.
Note: An empty query will result in listing of all the molecules present in the database.
Results
Phytochemical Search
Search by Phytochemical
Phytochemicals search is facilitated through various options. User can search a
phytochemical with its common name, for example,
‘Piperene’, its IUPAC name, ‘(2E,4E)-5- (1,3-benzodioxol- 5-yl)- 1-piperidin-
1-ylpenta-
2,4-dien-
1-one’ or its PubChem ID ‘638024’. The query will display the page for searched
phytochemical.
Users can use the ‘Explore’ tab to obtain full phytochemical profile. You may also
search
for
spice phytochemicals molecular weight, hydrogen bond donors/acceptors or partition
coefficient
(ALogP). If the user does not have any particular phytochemical to search, they can
perform
a
null search which will display a list of all available phytochemicals in the database.
Click here to view all the phytochemicals present in SpiceRx.
Phytochemical Profile Panel
This panel provides the complete ‘Phytochemical Profile’ of the molecule searched. This includes molecular identities such as MeSH ID, PubChem ID, IUPAC Name, Canonical and Isomeric SMILES and the Molecular Formula. Using JSmol applet, the panel also provides 2D image of the molecule, which transforms into 3D view upon clicking ‘View JSmol’ button. Importantly, using structural similarity search one could either explore similar molecules within SpiceRx, or could identify commercially available molecules from ZINC. Further, an option is provided to download the molecule data in Mol2, SDF, 2D image, and JSON format. Below the ‘Downloads’ button, clicking the ‘Spice/Herb’ button will provide a list of spices/herbs in which the phytochemical is present. ‘Disease’ button will provide the list of all diseases with which the given phytochemical is linked. Physicochemical as well as ADMET properties of the compound can also be obtained by clicking respective tabs.
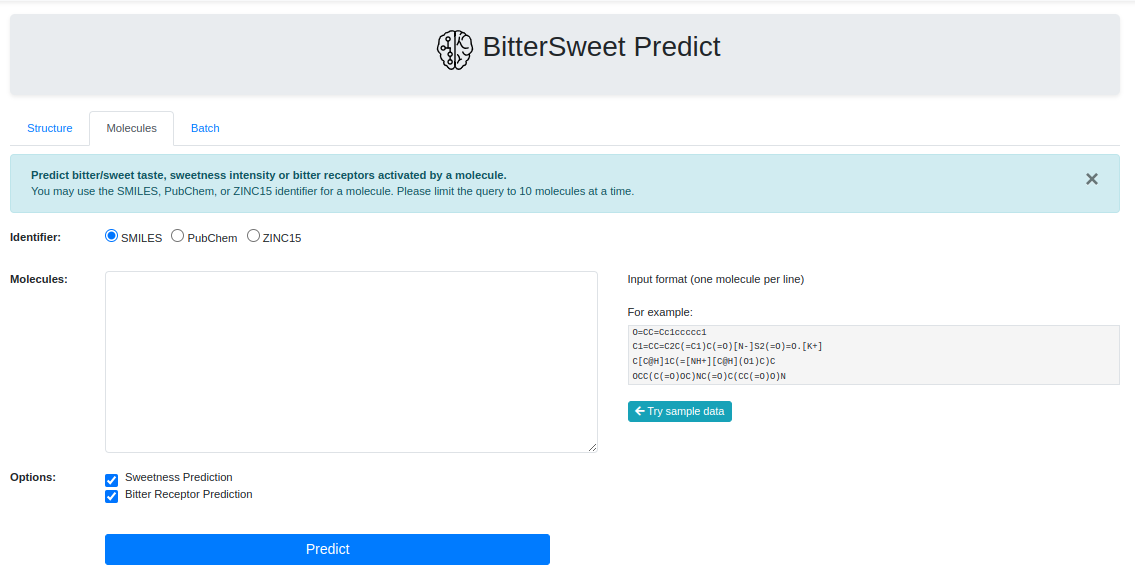
Prediction Tool
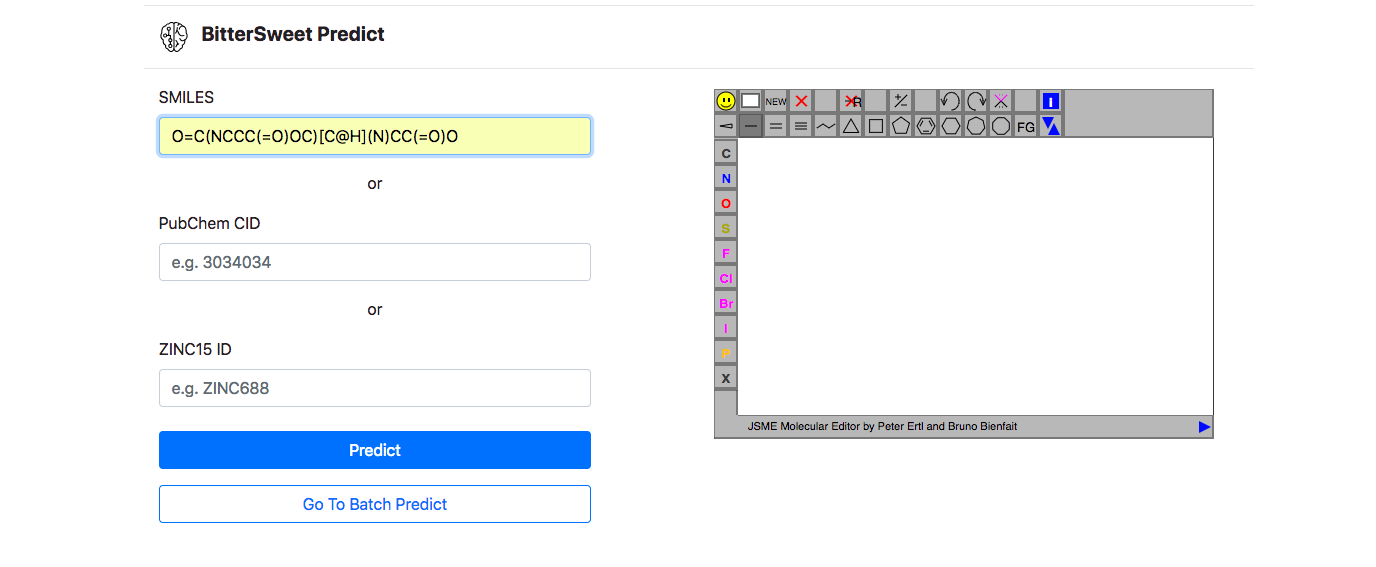
In addition to a comprehensive curation of Bitter-Sweet molecules, the resource provides with a tool for predicting the taste of a molecule on the BitterSweet axis ranging from Bitter, Bitter-Sweet, Neither Bitter nor Sweet, or Sweet by implementing state of the art machine learning classifiers. These models were trained from molecular properties obtained from ChemoPy - A freely available python package for calculating commonly used structural and physiochemical properties.
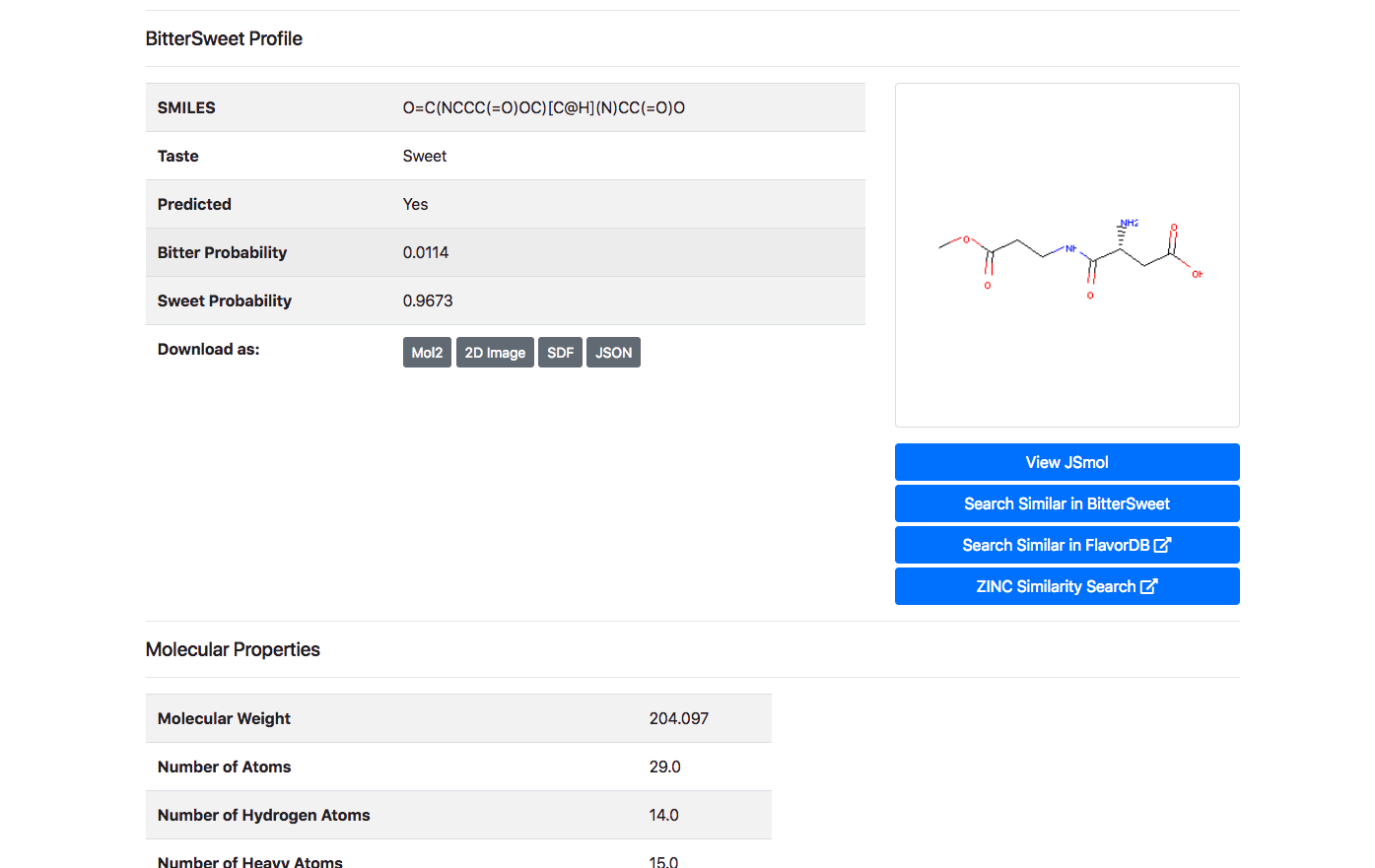
The following is a list of 10 sweet molecules of Mango from FlavorDB
which
you can yourself try
out
-
| SMILE ID | Name |
|---|---|
| CC1CCC(C(C1)O)C(C)C | Neomenthol |
| CC(=O)C(=O)C | 2,3-Butanedione |
| CCC1C(=O)C(=C(O1)C)O | 2-Ethyl-4-Hydroxy-5-Methyl-3(2H)-Furanone |
| CCCCCC(=O)C | 2-Heptanone |
| CCC(C)COC(=O)C | 2-Methylbutyl Acetate |
| CCCC(=O)C | 2-Pentanone |
| CCCC(=O)CC | 3-Hexanone |
| C1=CC=C(C=C1)CCC(=O)O | 3-Phenylpropanoic Acid |
| C(C1C(C(C(C(O1)OC2C(OC(C(C2O)O)O)CO)O)O)O)O | Alpha-Maltose |
| C1=CC=C(C=C1)CO | Benzyl Alcohol |
Reliability of Prediction Models
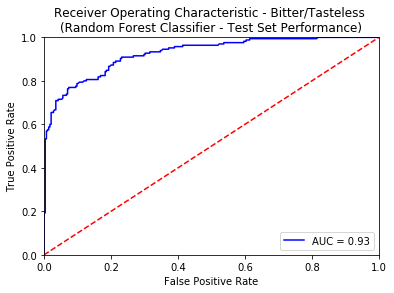
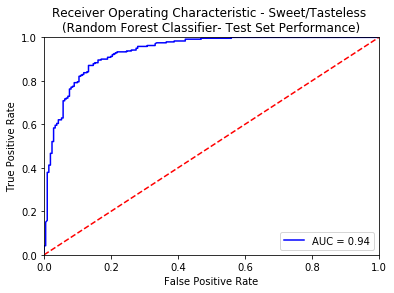
An ROC curve is the most commonly used way to visualize the performance of a binary classifier, and AUC is (arguably) the best way to summarize its performance in a single number. An area of 1 represents a perfect test; an area of .5 represents a worthless test. A rough guide for classifying the accuracy of a diagnostic test is the traditional academic point system:
- 0.90-1.00 = excellent
- 0.80-0.90 = good
- 0.70-0.80 = fair
- 0.50-0.70 = poor
- less than 0.50 = fail
Apart from using the standard tricks of avoiding overfitting (like Regularization, KFold Cross Validation), we also created an external validation set known as the Gold Standard from the molecules curated from 3 sources namely - Phyto Dictionary, BitterNew and UNIMI. A high AUC-ROC value of 0.87 on these molecules ensured the robustness of our classification models. For more details of the model training and comparative analysis of the state of the art, please refer to the data statistics dashboard.
Processing a Batch of SMILE Identifiers
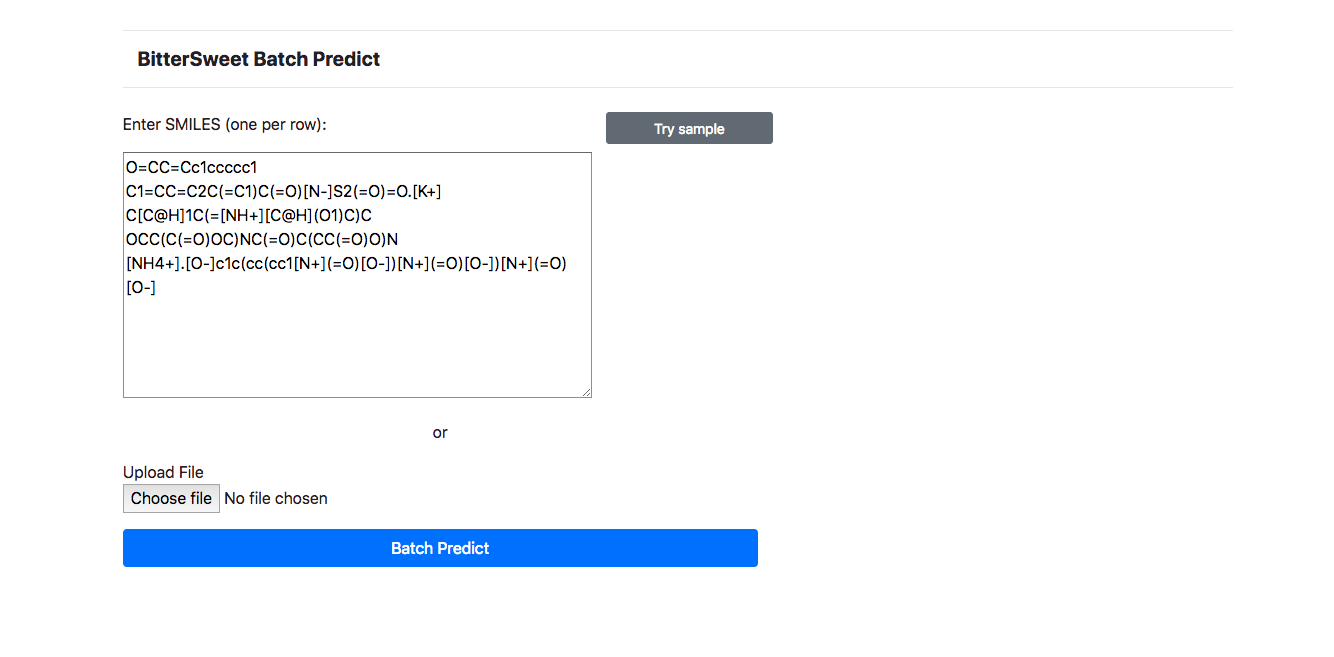
To further enhance the user experience and to improve the utility of the BitterSweet
Predict, Batch Prediction facility is provided for prediction of Bitter-Sweet
taste
on the basis of SMILE Identifiers. One may simply list a bunch of SMILES to fetch
predictions. To avoid inordinate delays, at the moment, Batch Predict allows
searching
upto 10 molecules.
Note: Batch Predictions may take a while to process since the properties of the
query molecules are generated dynamically in the backend and further processed by the
machine learning classifier. So please be patient while using this functionality.
Results
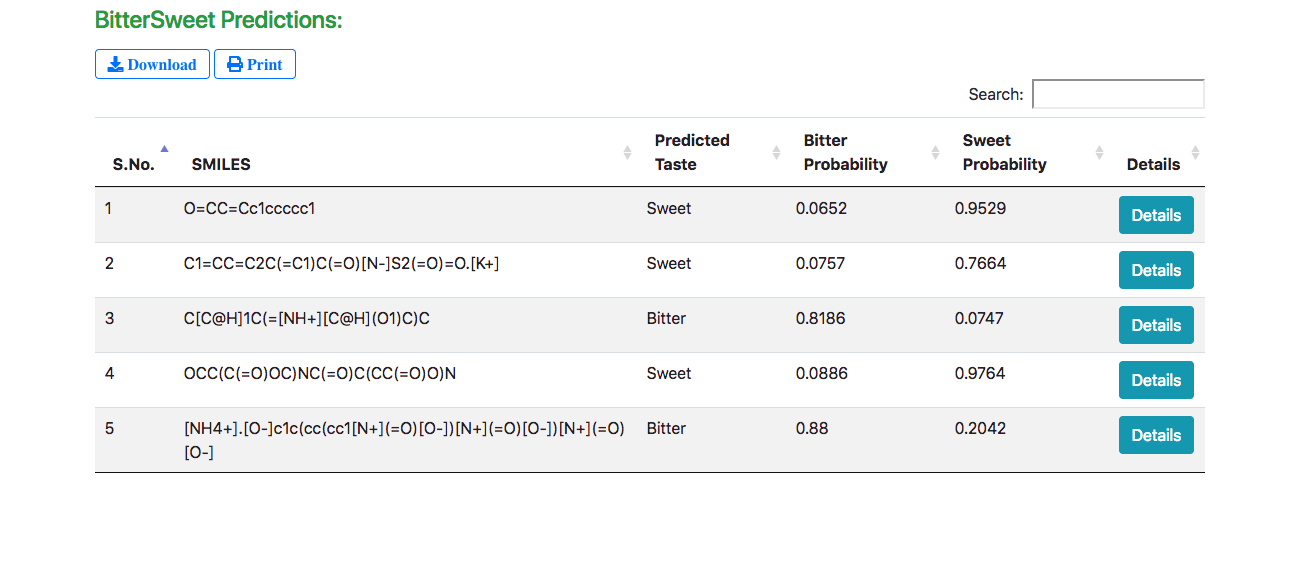
The results are displayed in a tabular form with the predicted taste, bitter and sweet confidence levels (probabilities) for every query molecule. The “Details” button enables exploring detailed molecular properties.
After entering a compound identifier and clicking the “Predict” button, BitterSweet starts processing the molecule based to generate all the chemical properties and to implement the classifiers at the back-end.This process may take a while (up to a minute, depending on the speed of your connection). Please be patient. After successful completion of the above steps, you will be redirected to the page detailing the predicted taste of the molecule.
Bitter and Sweet Probability
The probability of this molecule being bitter as predicted by the classifier. This value indicates the confidence level of the classifier. In simple terms the probability indicates, “How sure/unsure is the model that a given molecule is bitter or sweet?”. By default, as in case of any linear classifier, the threshold probability to categorize a class as True or False is set at 0.5, meaning that if the classifier predicts the probability of a certain class to be higher than 50%, it said to be truly belonging to that class. To demonstrate the robustness of our classifiers, herein we provide the False-Positive-Rate of our model against a range of threshold values.
| False Positive Rate - Bitter | Threshold Value - Bitter | False Positive Rate - Sweet | Threshold Value - Sweet |
|---|---|---|---|
| 0 | 0.98 | 0 | 1 |
| 0 | 0.87 | 0 | 0.98 |
| 0 | 0.74 | 0.01 | 0.97 |
| 0.01 | 0.67 | 0.02 | 0.94 |
| 0.06 | 0.5 | 0.03 | 0.86 |
| 0.09 | 0.46 | 0.06 | 0.77 |
| 0.15 | 0.42 | 0.09 | 0.71 |
| 0.2 | 0.35 | 0.12 | 0.62 |
| 0.27 | 0.28 | 0.16 | 0.5 |
| 0.35 | 0.21 | 0.21 | 0.44 |
| 0.47 | 0.15 | 0.28 | 0.37 |
| 0.6 | 0.09 | 0.41 | 0.23 |
| 0.7 | 0.05 | 0.59 | 0.14 |
| 0.82 | 0.02 | 0.82 | 0.05 |
Molecular Properties
These are chemical properties generated using ChemoPy open source library for python and portray various characteristics properties of a molecule. .